TranceptEVE: Combining Family-specific and Family-agnostic Models of Protein Sequences for Improved Fitness Prediction
Abstract
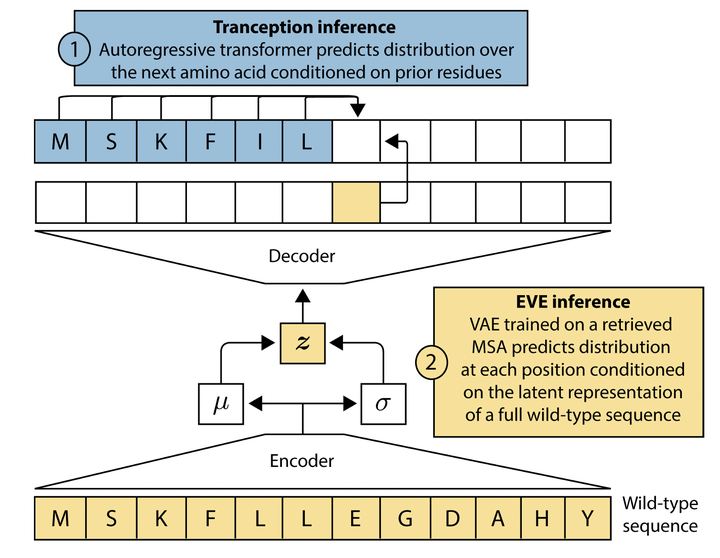
Modeling the fitness landscape of protein sequences has historically relied on training models on family-specific sets of homologous sequences called Multiple Sequence Alignments. Many proteins are however difficult to align or have shallow alignments which limits the potential scope of alignment-based methods. Not subject to these limitations, large protein language models trained on non-aligned sequences across protein families have achieved increasingly high predictive performance – but have not yet fully bridged the gap with their alignment-based counterparts. In this work, we introduce TranceptEVE – a hybrid method between family-specific and family-agnostic models that seeks to build on the relative strengths from each approach. Our method gracefully adapts to the depth of the alignment, fully relying on its autoregressive transformer when dealing with shallow alignments and leaning more heavily on the family-specific models for proteins with deeper alignments. Besides its broader application scope, it achieves state-of-the-art performance for mutation effects prediction, both in terms of correlation with experimental assays and with clinical annotations from ClinVar.